With this entry we conclude the series of posts where we have delved into the peculiarities of the different types of ELISA. After knowing the rationale and procedure on which each of them is based and seeing the main characteristics that differentiate them , today...
For all medicines and therapeutic agents , the WHO (World Health Organization) assigns a generic name known as INN ( International Non- proprietary Name ) in order to facilitate the identification of the active pharmaceutical ingredients that comprise it. This INN is also applicable in the case of monoclonal antibodies , which are assigned...
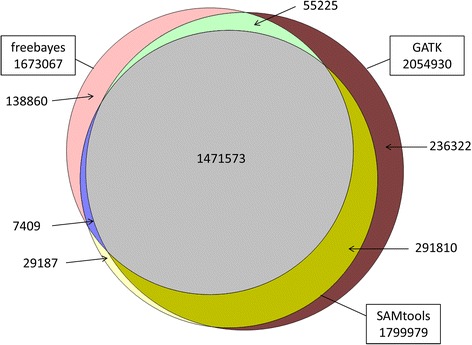
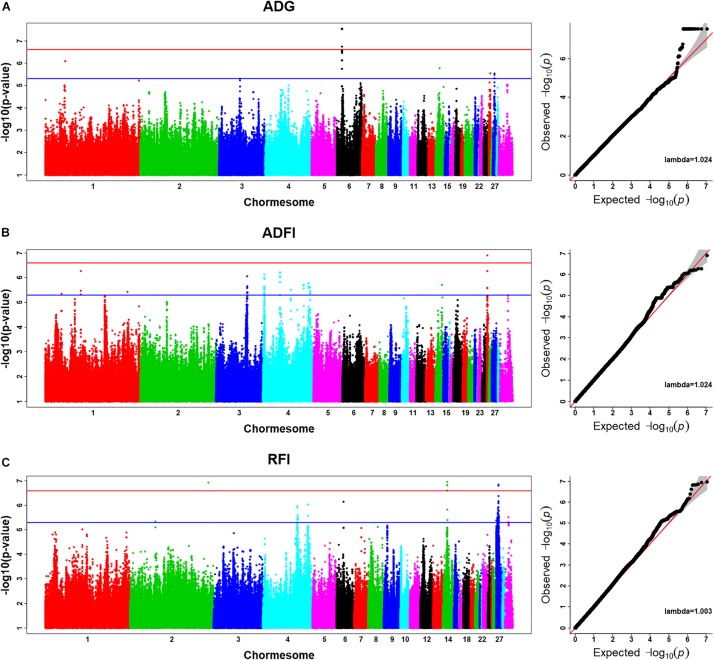
BACKGROUNDThe technical progress in the final decade has made it attainable to sequence tens of millions of DNA reads in a comparatively quick time-frame. Several variant callers primarily based on totally different algorithms have emerged and have made it attainable to...
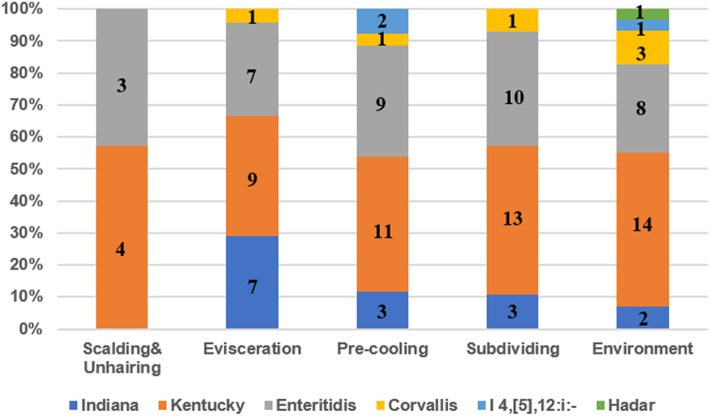
Salmonella has been referred to as a very powerful foodborne pathogen, which might infect people through consuming contaminated meals. Chicken meat has been referred to as an essential car to transmit Salmonella by the meals provide chain. This examine decided the prevalence, antimicrobial resistance,...
Poultry feed constitutes the most important price in poultry manufacturing, estimated to be as much as 70% of the whole price. Moreover, there’s strain on the poultry business to extend manufacturing to fulfill the protein demand of people and concurrently scale...
At the Gene Mapping Workshop held during the 22nd Conference of the International Society for Animal Genetics (ISAG) held in East Lansing, Michigan, USA it was resolved unanimously that animal gene nomenclature should “follow the rules for human gene nomenclature, including...
ARKdb can be queried in a number of ways. The main ways of querying are by locus/marker, by published reference or by map. You can also query by clone or library, although these tend to be less well-used areas of the...
ARKdb arose initially from the needs of a project co-ordinated at Roslin Institute to map the pig genome. The original intention was to utilise the Jackson Laboratory’s then current mouseGBASE database design, and this was actually implemented for pigs, chickens and...