
Prevalence of Salmonella Isolates and Their Distribution Based on Whole-Genome Sequence in a Chicken Slaughterhouse in Jiangsu, China.
Salmonella has been referred to as a very powerful foodborne pathogen, which might infect people through consuming contaminated meals. Chicken meat has been referred to as an essential car to transmit Salmonella by the meals provide chain.
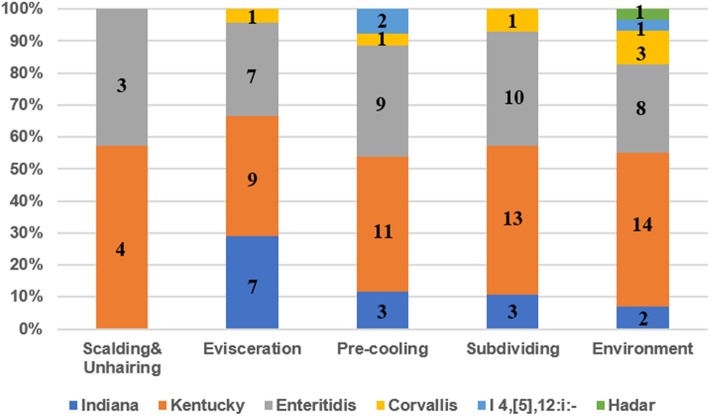
This examine decided the prevalence, antimicrobial resistance, and genetic traits of Salmonella at totally different rooster slaughtering levels in East China. In whole, 114 out of 200 (57%) samples had been Salmonella constructive, whereas Salmonella contamination was steadily rising from the scalding and unhairing stage (17.5%) to the subdividing stage (70%) all through the slaughtering.
Whole-genome sequencing (WGS) was then carried out to investigate the serotype, antimicrobial resistance gene profiles, and genetic relationship of all Salmonella isolates.
The most typical serotypes had been S. Kentucky (51/114, 44.7%) and S. Enteritidis (37/114, 32.5%), which had been distributed all through the 4 slaughtering levels, and had been additionally recognized in the corresponding environments.
The multilocus sequence typing (MLST) evaluation revealed that seven sequence varieties (STs) had been occupied by six totally different serotypes, respectively. Only S. Kentucky had two STs, ST314 was the predominant ST shared by 50 isolates, whereas the ST198 has 1 isolate. The antimicrobial resistance gene evaluation demonstrated that almost all of the strains belonging to S. Kentucky (39/51, 76.5%) and S. Indiana (15, 100%) contained over 5 teams of antimicrobial resistance genes. Based on the core genome evaluation, 50 S. Kentucky isolates had been genetically similar, indicating that one S. Kentucky pressure with the identical genetic background was prevalent in the rooster slaughtering line. Although 37 S.
Enteritidis isolates solely had three totally different antimicrobial resistance gene profiles, the core genome sequence evaluation subtyped these S. Enteritidis isolates into 5 totally different clusters, which revealed the varied genetic background of S. Enteritidis in the slaughterhouse.
The antimicrobial resistance phenotypes had been in step with the presence of the corresponding resistance genes of S. Kentucky and S. Enteritidis, together with tetA, floR, blaTEM-1B, strA/B, sul1/sul2, and gyrA (D87Y). Our examine noticed a excessive prevalence of Salmonella in the rooster slaughter line and recognized the slaughtering surroundings as a important supply of inflicting Salmonella cross-contamination throughout rooster slaughtering. Further research shall be wanted to restrict the transmission of Salmonella in the slaughterhouse.
Genome-Wide Detection of Key Genes and Epigenetic Markers for Chicken Fatty Liver.
Chickens are one of a very powerful sources of meat worldwide, and the incidence of fatty liver syndrome (FLS) is intently associated to manufacturing effectivity. However, the potential mechanism of FLS stays poorly understood.
An built-in evaluation of information from whole-genome bisulfite sequencing and lengthy noncoding RNA (lncRNA) sequencing was performed. A complete of 1177 differentially expressed genes (DEGs) and 1442 differentially methylated genes (DMGs) had been discovered. There had been 72% of 83 lipid- and glucose-related genes upregulated; 81% of 150 immune-related genes had been downregulated in fatty livers.
Part of these genes was inside differentially methylated areas (DMRs). Besides, sixty-seven lncRNAs had been recognized differentially expressed and divided into 13 clusters primarily based on their expression sample.
Some lipid- and glucose-related lncRNAs (e.g., LNC_006756, LNC_012355, and LNC_005024) and immune-related lncRNAs (e.g., LNC_010111, LNC_010862, and LNC_001272) had been discovered by a co-expression community and practical annotation.
From the expression and epigenetic profiles, 23 goal genes (e.g., HAO1, ABCD3, and BLMH) had been discovered to be hub genes that had been regulated by each methylation and lncRNAs.
We have supplied complete epigenetic and transcriptomic profiles on FLS in rooster, and the identification of key genes and epigenetic markers will increase our understanding of the molecular mechanism of rooster FLS.

Genetics and breeding of a black-bone and blue eggshell chicken line. 1. Body weight, skin color, and their combined selection

Transcriptome sequencing reveals genes involved in cadmium-triggered oxidative stress in the chicken heart
