
Comparison among three variant callers and assessment of the accuracy of imputation from SNP array data to whole-genome sequence level in chicken.
BACKGROUNDThe technical progress in the final decade has made it attainable to sequence tens of millions of DNA reads in a comparatively quick time-frame.
Several variant callers primarily based on totally different algorithms have emerged and have made it attainable to extract single nucleotide polymorphisms (SNPs) out of the whole-genome sequence.
Often, only some people of a inhabitants are sequenced utterly and imputation is used to receive genotypes for all sequence-based SNP loci for different people, which have been genotyped for a subset of SNPs utilizing a genotyping array.
METHODSFirst, we in contrast the units of variants detected with totally different variant callers, specifically GATK, freebayes and SAMtools, and checked the high quality of genotypes of the known as variants in a set of 50 totally sequenced white and brown layers. Second, we assessed the imputation accuracy (measured as the correlation between imputed and true genotype per SNP and per particular person, and genotype battle between father-progeny pairs) when imputing from excessive density SNP array data to whole-genome sequence utilizing data from round 1000 people from six totally different generations.
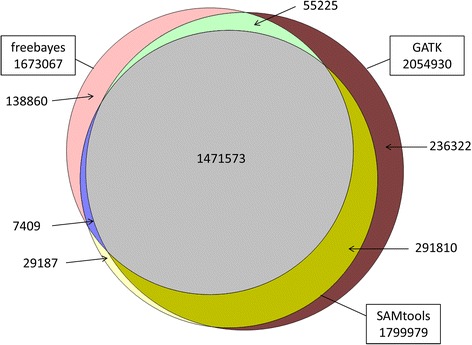
Three totally different imputation applications (Minimac, FImpute and IMPUTE2) have been checked in totally different validation eventualities.RESULTSThere have been 1,741,573 SNPs detected by all three callers on the studied chromosomes 3, 6, and 28, which was 71.6 % (81.6 %, 88.0 %) of SNPs detected by GATK (SAMtools, freebayes) in whole.
Genotype concordance (GC) outlined as the proportion of people whose array-derived genotypes are the identical as the sequence-derived genotypes over all non-missing SNPs on the array have been 0.98 (GATK), 0.97 (freebayes) and 0.98 (SAMtools). Furthermore, the share of variants that had excessive values >>0.9) for an additional three measures (non-reference sensitivity, non-reference genotype concordance and precision) have been 90 (88, 75) for GATK (SAMtools, freebayes).
With all imputation applications, correlation between unique and imputed genotypes was>>0.95 on common with randomly masked 1000 SNPs from the SNP array and>>0.85 for a leave-one-out cross-validation inside sequenced people.CONCLUSIONSPerformance of all variant callers studied was superb in normal, significantly for GATK and SAMtools.
FImpute carried out barely worse than Minimac and IMPUTE2 in phrases of genotype correlation, particularly for SNPs with low minor allele frequency, whereas it had lowest numbers in Mendelian conflicts in accessible father-progeny pairs. Correlations of actual and imputed genotypes remained consistently excessive even when people to be imputed have been a number of generations away from the sequenced people.
Genome-wide evaluation reveals the extent of EAV-HP integration in home hen.
BACKGROUNDEAV-HP is an historic retrovirus pre-dating Gallus speciation, which continues to flow into in trendy hen populations, and led to the emergence of avian leukosis virus subgroup J inflicting vital financial losses to the poultry trade.
We mapped EAV-HP integration websites in Ethiopian village chickens, a Silkie, Taiwan Country hen, purple junglefowl Gallus gallus and a number of inbred experimental strains utilizing whole-genome sequence data.RESULTSAn common of 75.22 ± 9.52 integration websites per fowl have been recognized, which collectively group into 279 intervals of which 5 % are frequent to 90 % of the genomes analysed and are suggestive of pre-domestication integration occasions.
More than a 3rd of intervals are particular to particular person genomes, supporting energetic circulation of EAV-HP in trendy chickens. Interval density is correlated with chromosome size (P < 2.31(-6)), and 27 % of intervals are situated inside 5 kb of a transcript.
Functional annotation clustering of genes reveals enrichment for immune-related capabilities (P < 0.05).CONCLUSIONSOur outcomes illustrate a non-random distribution of EAV-HP in the genome, emphasising the significance it might have performed in the adaptation of the species, and present a platform from which to lengthen investigations on the co-evolutionary significance of endogenous retroviral genera with their hosts.

Exploring the genetic architecture of feed efficiency traits in chickens
Genetic relatedness of multidrug resistant Escherichia coli isolated from humans, chickens and poultry environments
